Global Regulatory Development & Medical Writing
Extensive experience covering pre-marketing and post-marketing regulatory strategies for a wide-range of medical therapeutics, including drugs and biologics worldwide
Extensive experience covering pre-marketing and post-marketing regulatory strategies for a wide-range of medical therapeutics, including drugs and biologics worldwide
Accelerate Approvals with Experienced Regulatory Strategic Leadership and Execution
Managing today’s complex and ever-evolving regulatory demands requires strategic and expert insight. Drive the development of your products – from preclinical proof-of-concept candidates to approved products primed for post-marketing label extension and regulatory maintenance – by partnering with an experienced partner who knows how to help you reach your long-term commercial goals.


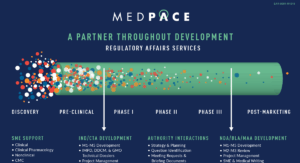
Global Regulatory Affairs – A Trusted Advisor
throughout Clinical Development
The Global Medpace Regulatory Affairs team works collaboratively with Sponsors on a national and local basis to help accelerate the development of safe and effective medical therapeutics. Our team has extensive experience covering pre-marketing and post-marketing regulatory strategies for a wide range of medical therapeutics, including drugs and biologics worldwide.
Global Regulatory Services
As a full-service regulatory partner, our strategic and operational global services include support for:
Medical Writing
Medpace’s dedicated team of medical writers hold advanced degrees ranging from PharmD to MDs, with background in Medpace’s therapeutic areas. Our team can support your clinical trial documentation needs by developing protocols, protocol amendments, and clinical study reports from early phase to post-approval. Our medical writing services include:
- Investigational and marketing applications
- Summary document preparation
- Protocols
- Clinical Study Reports (CSRs)
- Investigational medicinal product
- Dossier (IMPDs)
Strategic Development
Leverage our regulatory expert’s extensive experience to navigate the final steps of your pre-clinical program, transition to clinical development, and advance to pivotal clinical studies and market authorization.
- Gap analysis and target product profile (TPP) development
- Clinical synopsis development
- Evaluation of clinical trial design and endpoints
- Fast Track and PRIME
- Breakthrough, RMAT and QIDP
- Orphan drug applications
- Pediatric plans
- Accelerated and conditional approvals
Document Preparation & Submission
Our global regulatory experts conduct electronic publishing of regulatory documents and dossiers in eCTD format while facilitating submission to regulatory agencies via electronic gateways.
- Overview and summary document preparation
- e-CTD preparation, compilation, and e-publishing
- Electronic submission to regulatory authorities
- Submission maintenance and regulatory compliance
Regulatory Agency Interactions
Global regulatory affairs accelerates the global development of drugs, biological products and medical devices, by applying our experience of working with global and local regulatory authorities, and in-depth knowledge of the dynamic regulatory landscape.
- Strategy and planning
- Meeting requests and briefing documents
- Meeting rehearsals and facilitation
- Meeting minutes and follow-ups

Working with Us – Delivering Regulatory Strategies and Operational Support for Clinical Development
Global Regulatory Affairs provides scientifically-based, therapeutically-focused regulatory services from strategy to execution. Our team of global regulatory experts works with pharmaceutical, biotechnology, and medical device companies of all sizes, from early-stage companies to established companies.
Our teams are comprised of global product development experts, strategists, regulatory project managers, regulatory writers, and regulatory operations colleagues, providing unrivaled regulatory support across all areas of clinical development through early discovery to marketing authorization.